
Pablo García-Fernández personal webpage
Researcher and professor at Universidad de Cantabria (Spain)

Second-Principles Density Functional Theory
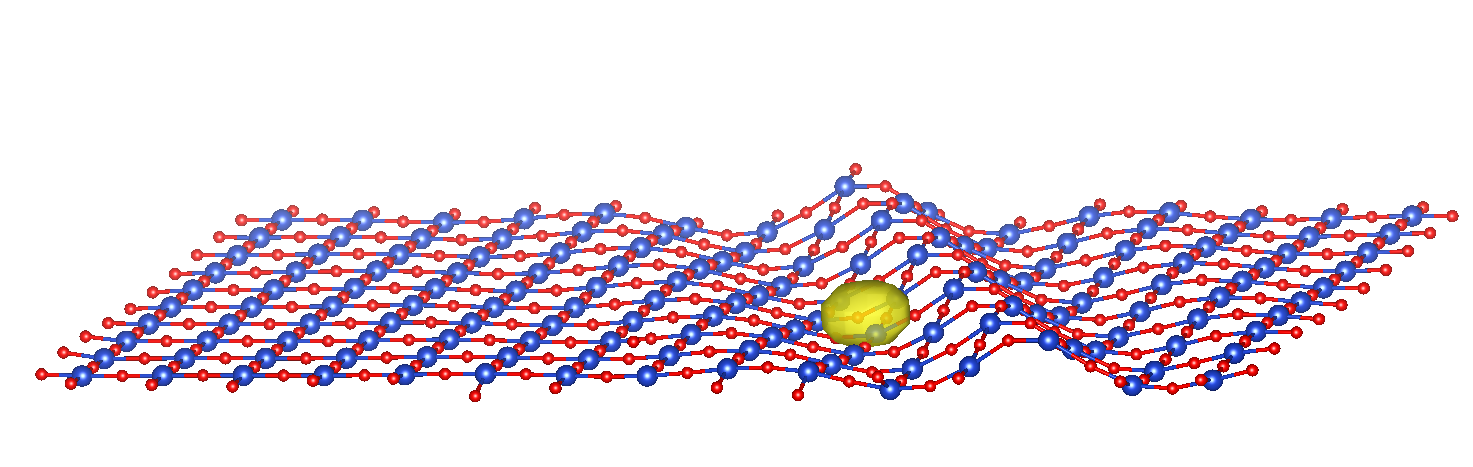
The SPDFT approach is a large-scale electronic structure method, prepared to deal with 100000+ atoms, based on a systematical approximation of the full DFT energy and with a similar level of accuracy. This is achieved by separating the total density into a reference part, typically corresponding to the system’s neutral, geometry-dependent ground state, and a deformation contribution that takes into account the difference between the full density and the reference one.
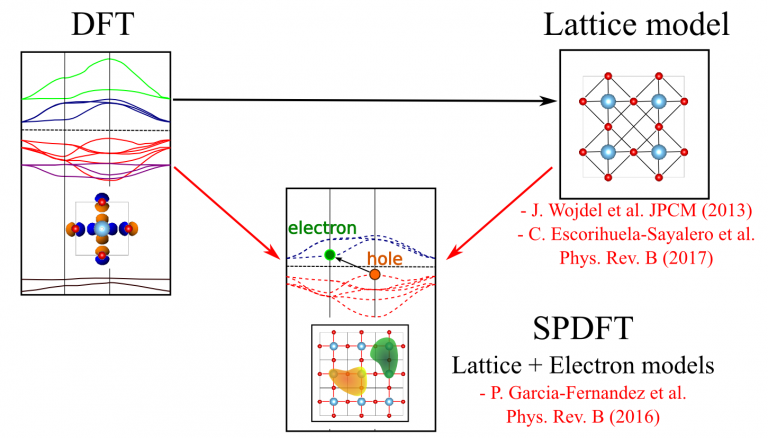 Expanding the DFT energy in terms of the deformation density it is found that the zeroth-order term, corresponding to the DFT energy associated to the reference density, simply describes, in most cases, the energy surface of the unperturbed ground state. Higher order terms describe electron-hole excitations that allow obtaining the full range of electron properties like band, magnetic states, etc.
Expanding the DFT energy in terms of the deformation density it is found that the zeroth-order term, corresponding to the DFT energy associated to the reference density, simply describes, in most cases, the energy surface of the unperturbed ground state. Higher order terms describe electron-hole excitations that allow obtaining the full range of electron properties like band, magnetic states, etc.
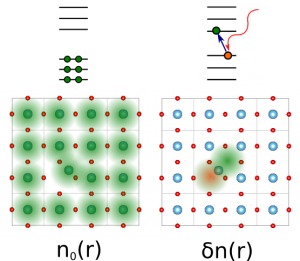 At difference with many other methods that are atom-based SPDFT is material -based. With this we mean that the basic construction block of a simulation is a model for a material. In particular, a SPDFT simulation requires the use of both a force-field and an electron Hamiltonian, formulated in a Wannier-function basis, that are obtained from first-principles simulations of a material in contrast with atom-atom potentials or electron basis based on atomic-functions typically used in other large-scale approaches. While this imposes the most important constraint to the method, the requirement of having a fixed bond topology – i.e. atoms cannot dissociate – this also allows the method to essentially match the accuracy of DFT even when dealing with very large number of atoms/electrons.
At difference with many other methods that are atom-based SPDFT is material -based. With this we mean that the basic construction block of a simulation is a model for a material. In particular, a SPDFT simulation requires the use of both a force-field and an electron Hamiltonian, formulated in a Wannier-function basis, that are obtained from first-principles simulations of a material in contrast with atom-atom potentials or electron basis based on atomic-functions typically used in other large-scale approaches. While this imposes the most important constraint to the method, the requirement of having a fixed bond topology – i.e. atoms cannot dissociate – this also allows the method to essentially match the accuracy of DFT even when dealing with very large number of atoms/electrons.